The pressure issue arose from some very unexpected behavior. When initially adding D2 at room temperature, it seemed to be absorbed within a matter of seconds, never reaching the target of 300 Pa. Once I noticed this, I closed the valves, with pressure at ~225 Pa. A video clip of this loading process will be posted soon.
After about 10 minutes settling time the pressure seemed stable, so we started to warm the reactor. At around 100°C, the pressure rose quickly to around 1000 Pa and continued to rise as the temperature increased. In the final power step it reached over 5000 Pa, far more than could be explained by the D2.
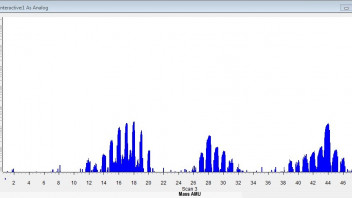
At the end of the last temperature step, we sampled the cell with the Cirrus mass spec. There was no deuterium seen in the cell, none detectable anyway. The plentiful gas at 5k Pa did show a range of masses suggesting compounds of carbon, oxygen and nitrogen, with deuterium atoms attached. Perhaps someone familiar with catalytic chemistry at 100-250°C could find some sense in this. The important question is if the rapid pressure rise starting at 100°C was due to abnormal desorbtion of introduced D2, or from a leak that didn't appear during de-gassing the system at room temperature. The cell reached around 5E-5 Torr before heating, so any leak path must be highly temperature-sensitive.